

2024捷报连连!集萃药康自主产权阿尔茨海默症小鼠模型FAD4T第四篇高质量文章发表!
2024年1月23日,安徽医科大学陈明课题组和浙江大学医学院唐斌梁课题组,与集萃药康研发科学家合作在Life Sciences(IF = 6.78)期刊上在线发表了题为“Cognitive impairment in Alzheimer's disease FAD4Tmouse model: Synaptic loss facilitated by activated microglia via C1qA ”的论文。该研究使用了来自集萃药康自主产权阿尔茨海默病(Alzheimer’s disease, AD)小鼠模型FAD4T(C57BL/6JGpt-Tg (Thy-APP/Thy-PSEN1) 5/Gpt,T053302)作为AD认知障碍小鼠模型,发现了与认知功能障碍相关的一系列分子变化。通过行为测试和免疫荧光染色技术,研究人员观察到了FAD4T小鼠模型脑中Aβ斑块的存在以及大脑皮层和海马区域小胶质细胞的异常激活,进一步验证了认知损伤的发生;通过海马神经元的膜电生理记录和Golgi染色技术,研究人员发现了突触后电流幅度的降低以及突触的形态异常,提示FAD4T小鼠模型的突触功能受损;进一步的RNA-seq分析揭示了免疫系统和促炎基因的表达升高,特别是C1qA蛋白和mRNA水平的增加,这表明过度激活的小胶质细胞可能是导致神经元突触丢失的关键因素。总的来说,这项研究提供了对AD患者认知损伤机制的深入理解,并初步探讨了针对C1qA的潜在治疗方法。这一研究有望为未来开发更有效的治疗策略提供重要的指导,并为我们更好地理解和应对AD这一挑战性疾病提供了新的视角[1]。

先前的研究表明,过度激活胶质细胞,特别是围绕Aβ斑块的小胶质细胞和星形胶质细胞,导致神经元凋亡,进而导致记忆丧失和认知功能障碍[2,3]。此外,小胶质细胞的吞噬在突触丢失中起着至关重要的作用[4,5,6]。小胶质细胞通过吞噬过多的突触来微调神经环路,这一过程部分依赖于补体介导的突触修剪。具体来说,作为经典补体途径的启动子,C1q由脑中的小胶质细胞产生,与突触标记共定位,并可能在各种神经退行性模型中促成突触丢失[7,8,9],由补体活性导致的突触丢失机制仍需要深入研究。因此,本项研究利用AD模型之一——FAD4T小鼠模型,进一步从不同层面对该机制进行了细致研究。
作者首先确定了FAD4T小鼠的病理表型和行为学障碍后,利用高尔基染色和WB分别对树突棘的密度和突触后支架蛋白PSD-95的表达进行探究。结果表明,在FAD4T小鼠的皮层和海马中,树突棘的密度均显著降低(图1 A-F),海马中PSD-95的表达也显著减少(图1 G-H)。树突棘作为接收突触前信号的关键结构,其数量和形态对突触功能至关重要。因此接下来,作者又利用膜片钳技术来记录突触后电流的变化。结果发现,与对照组相比,FAD4T小鼠的海马mEPSC的幅度显著降低,表明该模型小鼠的兴奋性突触传递受损(图2 A-F)。紧接着,作者又量化了兴奋性突触后受体的表达,发现在FAD4T小鼠的海马中,介导兴奋性突触后功能之一的NMDA受体亚基NMDAR1显著减少(图2 G-J)。这些结果共同表明FAD4T小鼠的突触后功能异常。
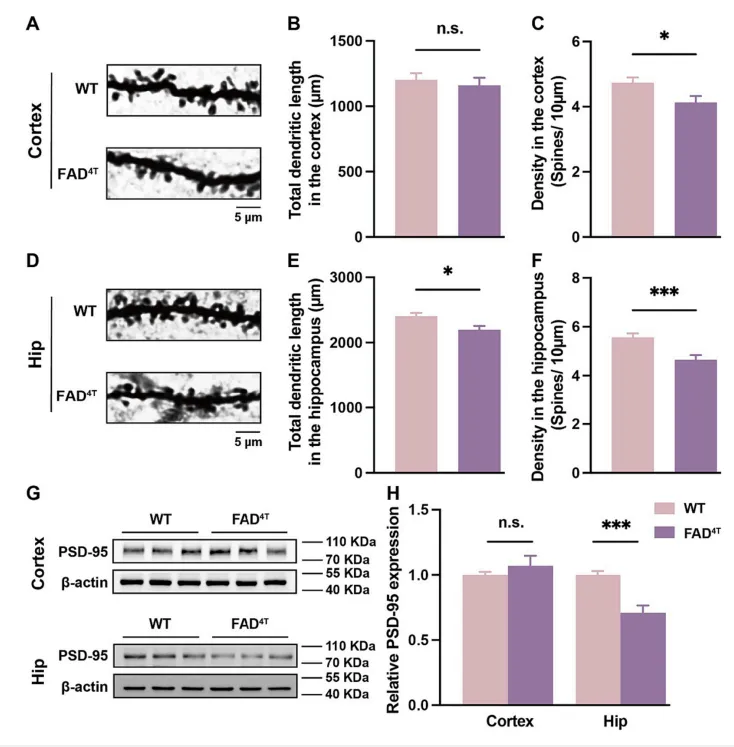
图1.FAD4T小鼠表现出异常的突触丢失
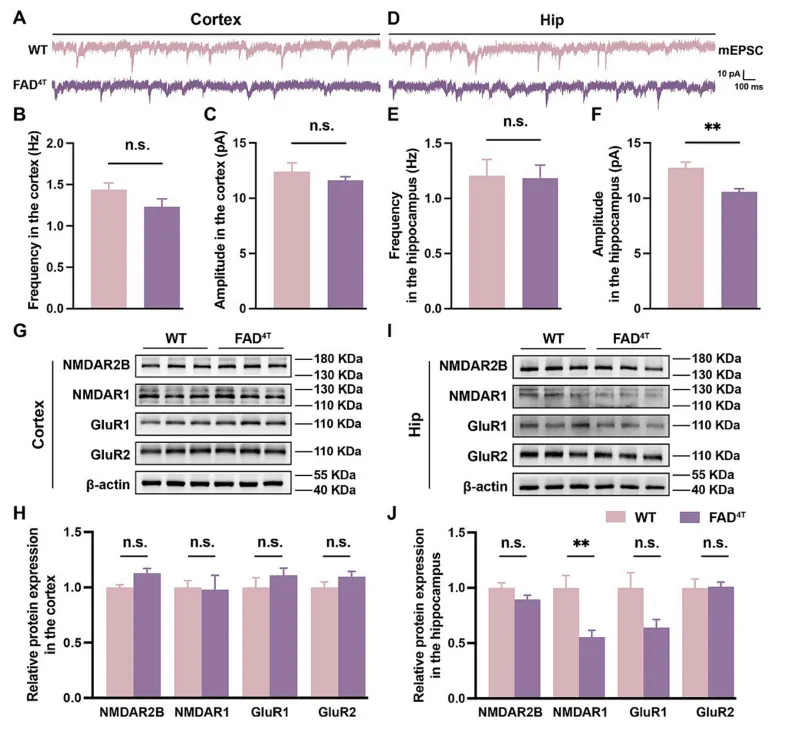
图2. FAD4T小鼠海马mEPSC振幅和兴奋性突触后受体表达降低
为了进一步探究FAD4T小鼠认知功能障碍的潜在发病机制,作者对WT和FAD4T小鼠的皮层和海马进行了RNA-seq分析。结果发现,在FAD4T小鼠的海马和皮层中,有很多差异表达的基因(DEGs)。其中许多免疫系统相关基因(如Ccl4和Ccl6)以及与AD发病和胶质细胞活化高度相关的基因(如Gfap、CD68、cst7和Trem2)表达均上调。此外,补体因子C1qA在两个组织中也明显上调(图3 A-E),且这些DEGs的产物在蛋白-蛋白相互作用网络中相互作用(图3F-G)。这些结果共同表明,FAD4T小鼠的小胶质细胞中的免疫系统,包括补体系统都被激活,而这也被认为是AD发病和发展的重要因素。
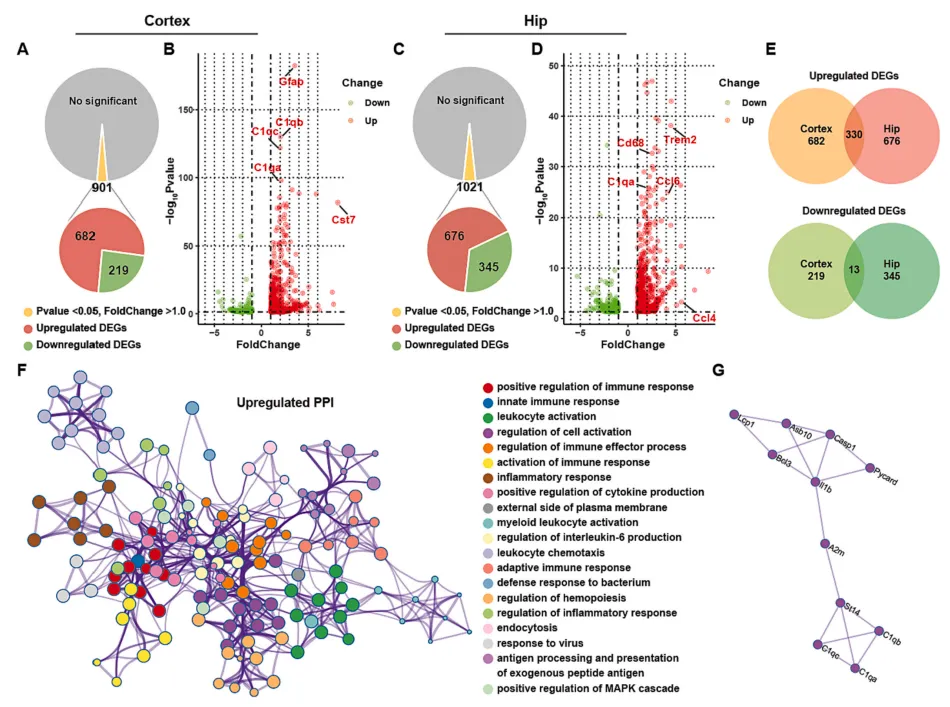
图3. RNA测序结果显示FAD4T小鼠中差异表达基因
C1q是主要在小胶质细胞中表达的经典补体通路的一个组成部分,而C1qA作为成员之一,与多种神经疾病相关。因此,接下来作者重点研究了C1qA对FAD4T小鼠神经炎症和突触功能的潜在影响。WB和qPCR的结果表明,FAD4T小鼠皮层和海马中C1qA蛋白和mRNA水平均显著升高(图4 A-F)。同时,免疫荧光染色显示FAD4T小鼠皮层和海马周围激活小胶质细胞中的C1qA浓度显著升高(图4 G-J)。此外,与WT小鼠相比,FAD4T小鼠皮层和海马组织中炎症因子--TNF-α和IL-18蛋白及mRNA表达水平均显著升高(图4 A-F)。这些结果表明补体成分C1qA有助于FAD4T小鼠神经炎症的加剧。
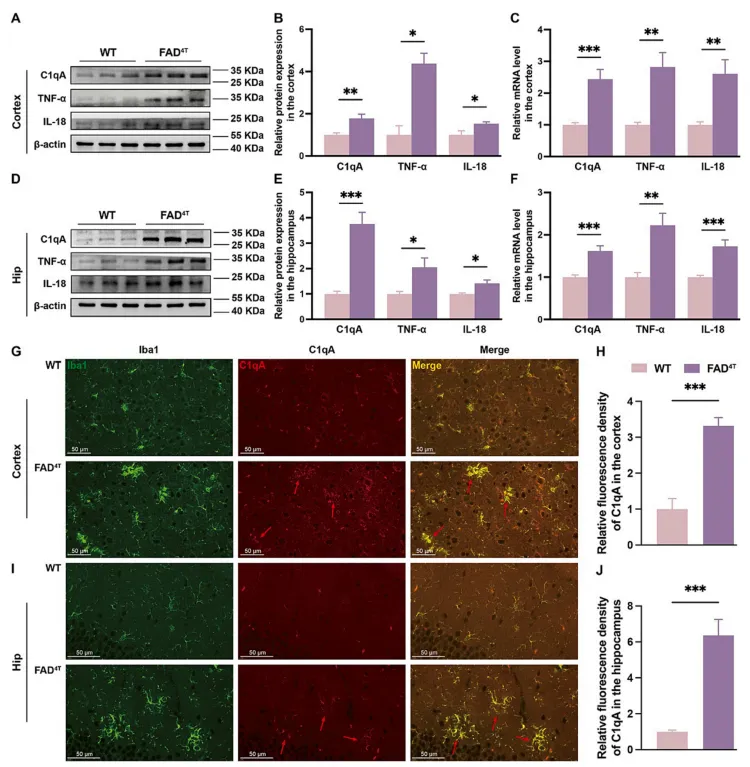
图4. FAD4T小鼠炎症因子TNF-α、IL-18及补体成分C1qA表达水平升高
有研究表明,催产素(Oxytocin,OT)作为一种主要负责分娩、结合和哺乳的激素肽,也参与调节啮齿动物中枢神经系统的认知行为。因此接下来作者在FAD4T小鼠模型中探究OT鼻腔给药是否可以恢复小鼠的认知和病理表型。给药7周后,水迷宫测试结果显示OT治疗后FAD4T小鼠的逃避潜伏期降低,FAD4T小鼠穿越平台次数和穿越总次数均增加,表明OT可改善FAD4T小鼠的认知缺陷(图5 A-H);免疫荧光结果显示,OT治疗组FAD4T小鼠的Aβ斑块大小和数量均减少,海马小胶质细胞数量明显减少(图6 A-C)。同时,对补体成分C1qA和炎症因子TNF-α、IL-1β和IL-18的定量研究显示,OT治疗可以显著降低FAD4T小鼠的C1qA和炎症相关成分的表达(图6 D-J)。这些结果都表明,OT可能通过降低C1qA的表达和降低Aβ斑块的产生来降低神经炎症水平并减轻突触缺陷,最终实现对FAD4T小鼠认知缺陷的改善。
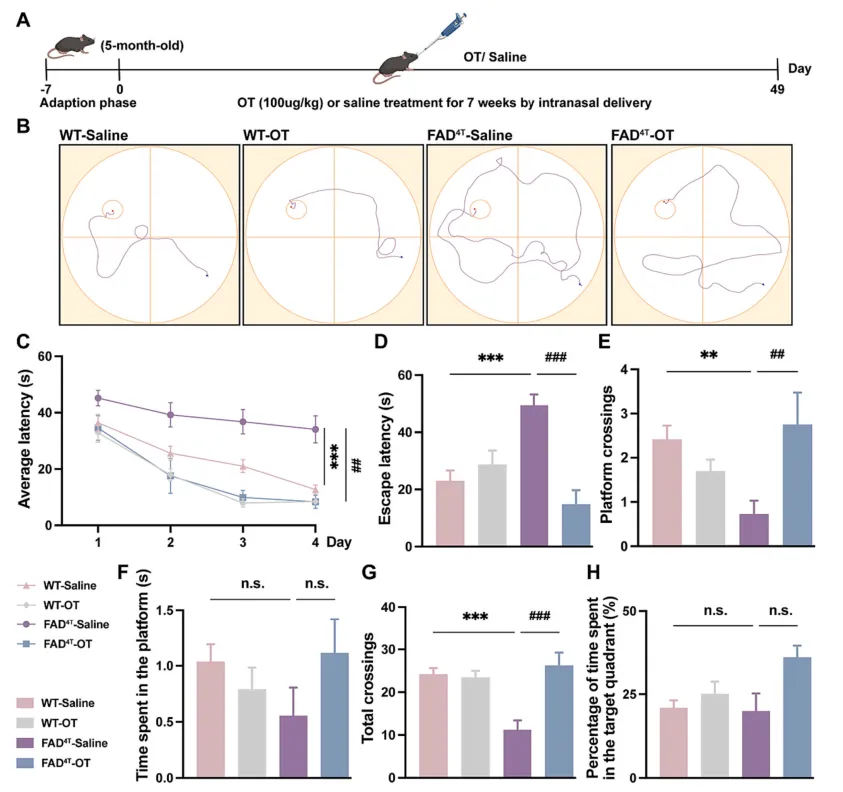
图5. OT治疗可改善FAD4T小鼠的认知障碍
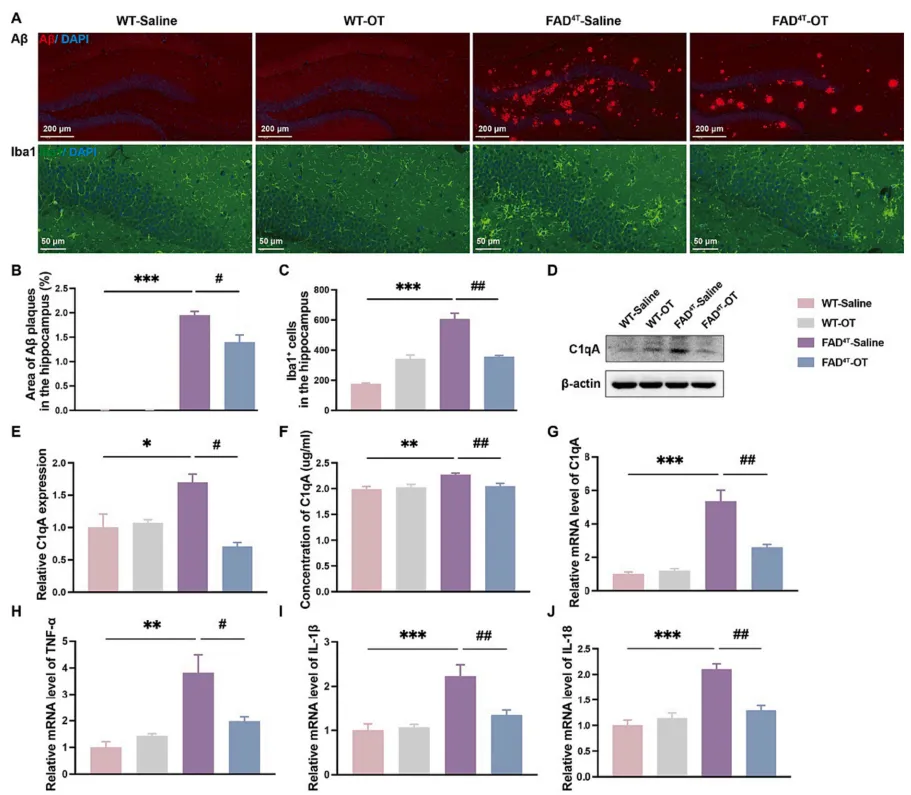
图6. OT治疗可减少FAD4T小鼠Aβ斑块和小胶质细胞数量,并通过C1qA减轻神经炎症
总结来说,该研究利用FAD4T小鼠作为AD模型鼠,发现:1) FAD4T小鼠表现出认知障碍并伴有轻度社交缺陷;2) FAD4T小鼠出现Aβ斑块沉积、神经胶质细胞活化、免疫活性上调等表型;3) FAD4T小鼠出现异常突触丢失和突触功能障碍;4)补体成分C1qA上调介导FAD4T小鼠神经炎症;5) OT可能通过抑制C1qA依赖性神经炎症来改善AD小鼠空间记忆缺陷(图7)。
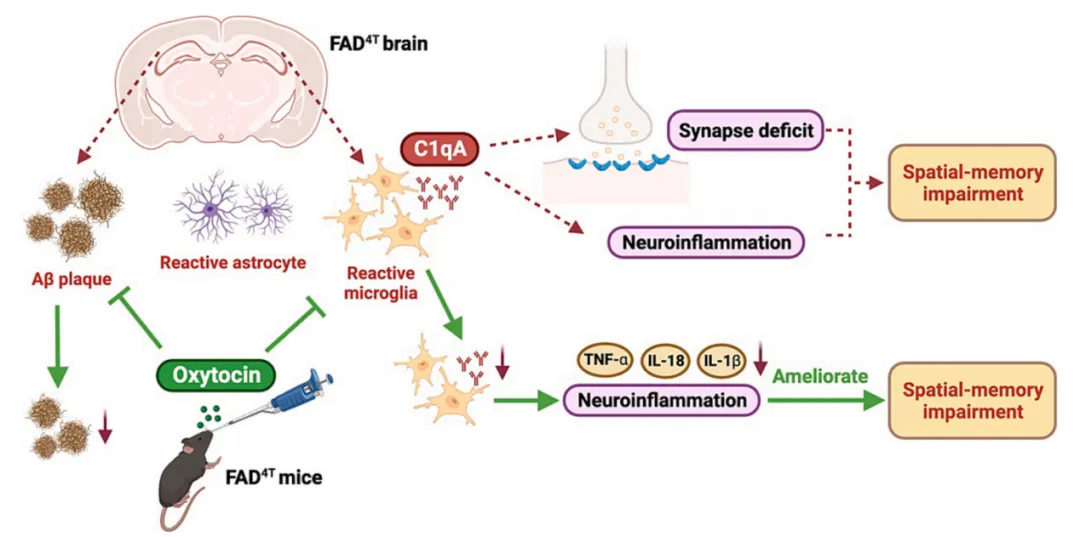
图7. OT调节C1qA介导的FAD4T小鼠突触功能和空间记忆的机制模式图
集萃药康自主知识产权FAD4T模型小鼠,拥有携带Swedish、Indiana突变的人源APP基因和携带M146L、L286V突变的人源PSEN1基因,可以很好的模拟临床AD患者的疾病表征,也能够满足相关药效需求。目前已有4篇相关文章发表,更多该模型相关文章在投稿或审核中,相信后续会有更多的高分文章与大家见面。除此模型之外,集萃药康还拥有FAD3T、FAD2T及以FAD4T为基础构建的携带人源化靶点(如hTREM2等)的AD Plus小鼠模型已经上市。如需了解更多模型详情,欢迎联系您身边熟悉的销售同事或致电官方客服电话400-966-0890获取详细数据资料。
参考文献:
[1] Zhang C, et al., Cognitive impairment in Alzheimer's disease FAD4T mouse model: Synaptic loss facilitated by activated microglia via C1qA. Life Sci. 2024 Jan 23;340:122457.
[2] M.S. Gee, et al., A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse, Alzheimers Res. Ther. 1 (2020) 45.
[3] L. Tao, et al., Microglia modulation with 1070-nm light attenuates Aβ burden and cognitive impairment in Alzheimer's disease mouse model, Light Sci Appl. 1 (2021) 179.
[4] S. Hong, et al., Complement and microglia mediate early synapse loss in Alzheimer mouse models, Science 6286 (2016) 712–716.
[5] M.J. Vasek, et al., A complement-microglial axis drives synapse loss during virusinduced memory impairment, Nature 7608 (2016) 538–543.
[6] H. Lui, et al., Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation, Cell 4 (2016) 921–935.
[7] D.P. Schafer, et al., Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner, Neuron 4 (2012) 691–705.
[8] B. Stevens, et al., The classical complement cascade mediates CNS synapse elimination, Cell 6 (2007) 1164–1178.
[9] C.J. Bohlen, et al., Microglia in brain development, homeostasis, and neurodegeneration, Annu. Rev. Genet. (2019) 263–288.