

世界渐冻人日:夏至已至,集萃药康助力融化渐冻症
2024年6月21日迎来了世界的第24个“世界渐冻人日”。该节日的设立旨在提升公众对渐冻症的了解与关注。自1997年起,“渐冻人”协会国际联盟将每年的6月21日定为世界渐冻人日,这一天恰好是北半球一年中日照时间最长的一天,寓意着盛夏的阳光可以消融冰雪,为渐冻症患者带来希望。
渐冻症初相识
渐冻症,学名肌萎缩侧索硬化症(Amyotrophic Lateral Sclerosis, ALS),是一类由大脑和脊髓中运动神经元渐进性死亡引起的全球第三大神经退行性疾病。ALS的主要临床特征包括进行性肌肉无力、肌肉萎缩以及运动障碍,并引起瘫痪以及呼吸困难,最终导致死亡;病理检测表明患者大脑和脊髓的运动神经细胞退变,皮质脊髓束和皮质延髓束出现轴突变性和毒性蛋白聚集[1]。
ALS属于罕见病的一种,发病率较低,全球范围内每10万人中大约有4~5名渐冻症患者。ALS疾病的致病机制目前尚未明确,已确定的风险基因影响包括兴奋性毒性、氧化应激、线粒体功能紊乱等多个信号通路,涉及SOD1、TARDBP、FUS和C9ORF72等25个基因[2]。目前全球仅有3款小分子药物获批应用于ALS患者的治疗,分别是利鲁唑(调节谷氨酸浓度和作用于离子通道而发挥作用)、依达拉奉(自由基清除)和Relyvrio(缓解线粒体功能障碍和内质网应激),其中利鲁唑和依达拉奉的药物治疗效果并不明显,仅能微弱延长患者的生存期2-3个月左右[3, 4],Relyvrio这一复方制剂也由于三期实验并未取得明显改善效果而于今年选择主动撤市,难以为ALS患者带来实质性的临床获益。
针对ALS疾病的药物研发正处于如火如荼的阶段,已在临床阶段的在研药物中,包括了以间充质干细胞为代表的细胞疗法、反义寡核苷酸疗法(Antisense oligonucleotides, ASO)技术敲低基因表达的基因疗法以及各类单抗研究等。其中由Ionis开发并授权给渤健的ASO药物QALSODY™(Tofersen)已获FDA加速批准上市,用于治疗SOD1基因突变导致的成人肌萎缩性侧索硬化症[5]。近日,研究团队也公布了使用Toferson治疗渐冻症的突破性进展。研究结果显示,Toferson显著抑制了一位渐冻症患者的疾病进展,不仅延长了患者的生存期,并且在经过4年的治疗后,患者仍然可以完成爬楼梯等自主运动,并未对生活造成较大的影响。基因治疗给渐冻症患者带来了冰雪渐融、春日将近的希望。
集萃药康ALS小鼠模型助力新药研发
小鼠模型对于研究神经退行性疾病的发病机制和新药研发都具有十分重要的作用。针对ALS疾病发生的热门基因SOD1和TARDBP,集萃药康构建了拥有自主产权的B6-hSOD1 G93A,hSOD1小鼠模型以及B6-hTDP43wtxA315T小鼠模型,可应用于针对SOD1基因和TARDBP基因的ALS药理药效及发病机制的研究。
B6-hSOD1 G93A,hSOD1小鼠模型携带G93A突变的人源SOD1全长基因,可以很好地满足针对SOD1靶点的ASO药物和其他ALS药物的评价需求。目前已收集到的验证数据表明该小鼠雄鼠在5.5月龄左右发病,并表现出符合ALS临床特征的运动功能下降,适用于ALS药理药效及发病机制的研究。
1. B6- hSOD1 G93A, hSOD1小鼠蛋白表达水平验证
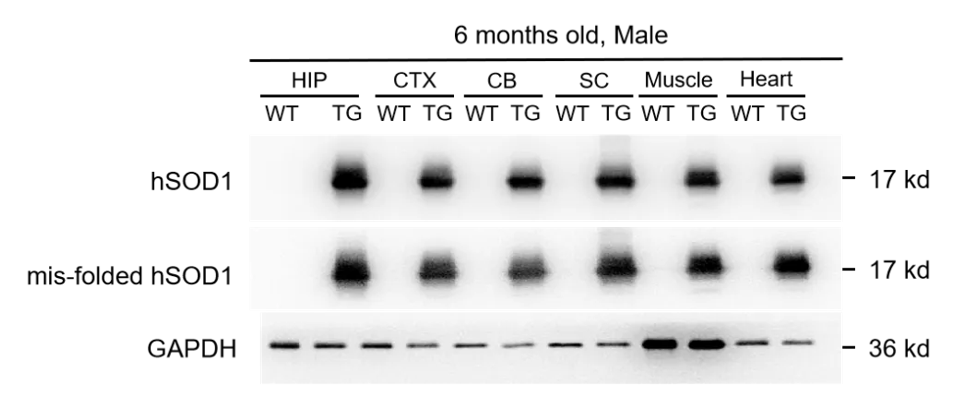
图1. B6-hSOD1 G93A, hSOD1小鼠蛋白表达水平验证
Western blot检测结果表明,B6-hSOD1 G93A, hSOD1转基因小鼠在脑、脊髓和肌肉中表达人源化SOD1蛋白,且会形成错误折叠的SOD1蛋白。(HIP: Hippocampus, CTX: Cortex, CB: Cerebellum, SC: Spinal cord)
2. B6- hSOD1 G93A, hSOD1小鼠肌肉萎缩表型
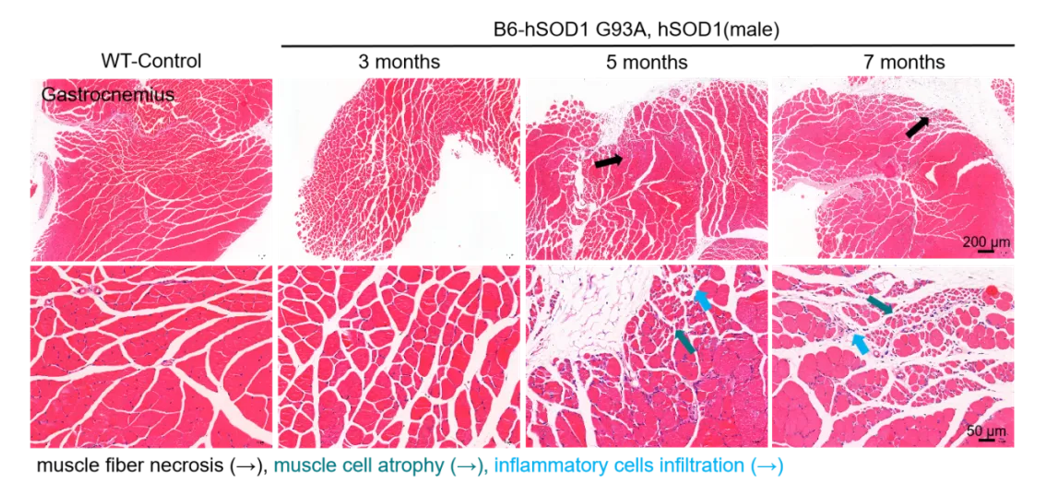
图2. B6-hSOD1 G93A, hSOD1小鼠肌肉病理染色
B6-hSOD1 G93A, hSOD1小鼠3月龄起可检测到腓肠肌的肌肉细胞变性坏死。肌纤维坏死(图中黑色箭头),肌细胞萎缩(图中绿色箭头),免疫细胞浸润(图中蓝色箭头)。
3. B6- hSOD1 G93A, hSOD1小鼠行为学数据显示雄鼠发病起始时间为5.5月龄左右
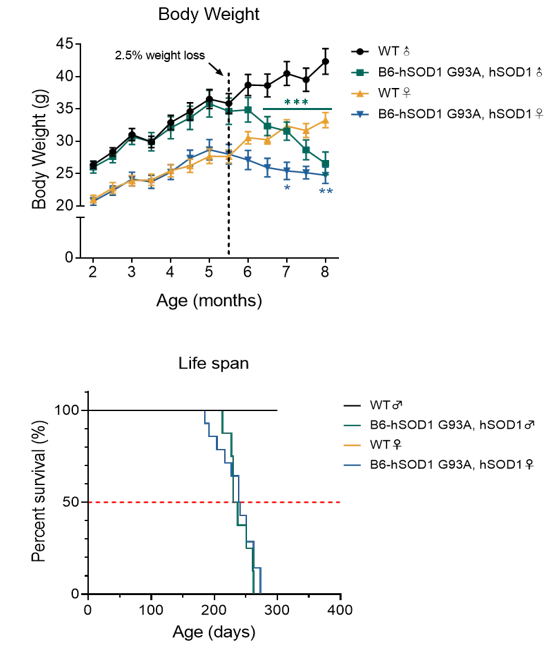
图3. B6-hSOD1 G93A, hSOD1小鼠体重变化及生存期曲线
与同窝阴性对照组相比,B6-hSOD1 G93A, hSOD1雄鼠5.5月龄起体重开始出现下降趋势,6.5月龄起体重出现显著性差异;小鼠生存期在270天左右。
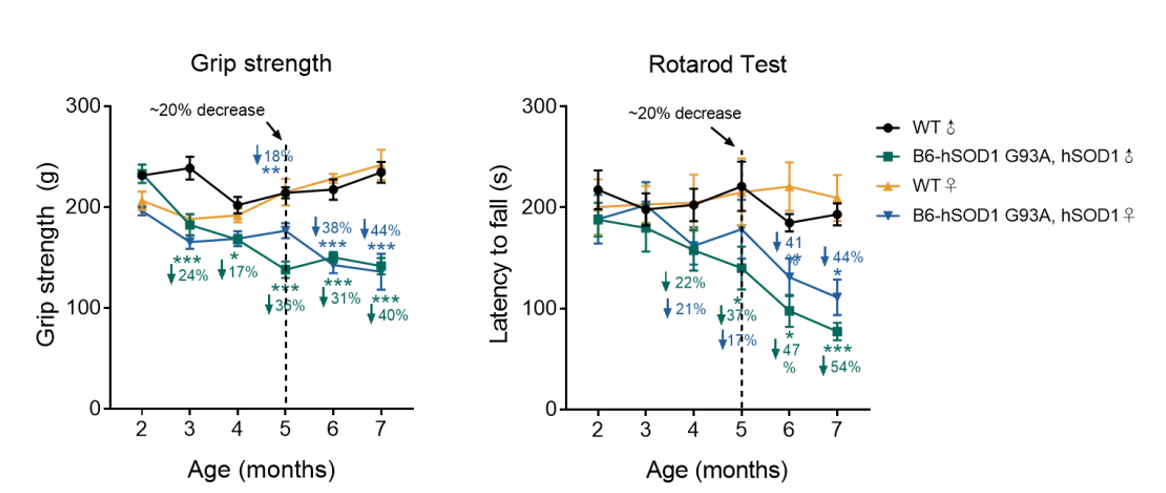
图4. B6-hSOD1 G93A, hSOD1小鼠运动能力变化
抓力测试实验结果显示,与同窝阴性对照组小鼠相比,B6-hSOD1 G93A, hSOD1雌雄鼠在5-5.5月龄开始抓力出现显著下降;转棒测试实验结果显示,B6-hSOD1 G93A, hSOD1雌雄鼠在5-5.5月龄起出现统计学显著性差异,提示小鼠运动协调性降低。
而另一个渐冻症模型B6-hTDP43wtxA315T小鼠模型携带A315T突变的人源TARDBP基因,可以满足针对TDP43靶点药物的评价需求。目前已收集到的验证数据表明该小鼠模型可部分模拟ALS病人病理及行为学特征,应用于ALS药理药效及发病机制的研究。
此外,我们也收集了两个模型的其他一些数据资料,例如运动神经元丢失、肌肉电生理、转录组学数据及神经免疫相关因子表达情况等。
如有需要,可联系销售经理或致电官方客服电话400-966-0890获取详细数据资料。
参考文献
1. Zhang F, Strom A L, Fukada K, et al. Interaction between Familial Amyotrophic Lateral Sclerosis (ALS)-linked SOD1 Mutants and the Dynein Complex. Journal of Biological Chemistry, 2007, 282(22):16691-16699.
2. Mead, R.J., Shan, N., Reiser, H.J. et al. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov 22, 185–212 (2023).
3. Jaiswal, M.K., Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev, 2019. 39(2): p. 733-748.
4. Chio, A., L. Mazzini, and G. Mora, Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology, 2020. 167: p. 107986.
5. Miller TM, Cudkowicz ME, Genge A, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387:1099-1110.