

破局罕见病困境:Alpl-KO小鼠助推HPP疗法开发
低磷酸酯酶症(HPP)是一种罕见的遗传性系统性疾病,由于合成碱性磷酸酶ALP的ALPL基因突变,导致组织非特异性碱性磷酸酶(TNSALP)活性缺失,引发骨骼矿化障碍以及神经递质耗竭。HPP临床表现较为多样,涵盖了骨骼畸形、钙磷代谢紊乱、反复骨折、呼吸衰竭、早发性牙齿脱落及新生儿致死。TNSALP作为定位细胞膜上的酶,能够催化无机焦磷酸盐(PPi)及5'-磷酸吡哆醛(PLP)代谢为下游物质,发挥骨骼矿化功能及合成维生素B6。当机体存在ALPL基因突变时,可导致TNSALP折叠、组装、调节或转运发生异常,造成胞外累积PPi以及PLP,如下图所示。
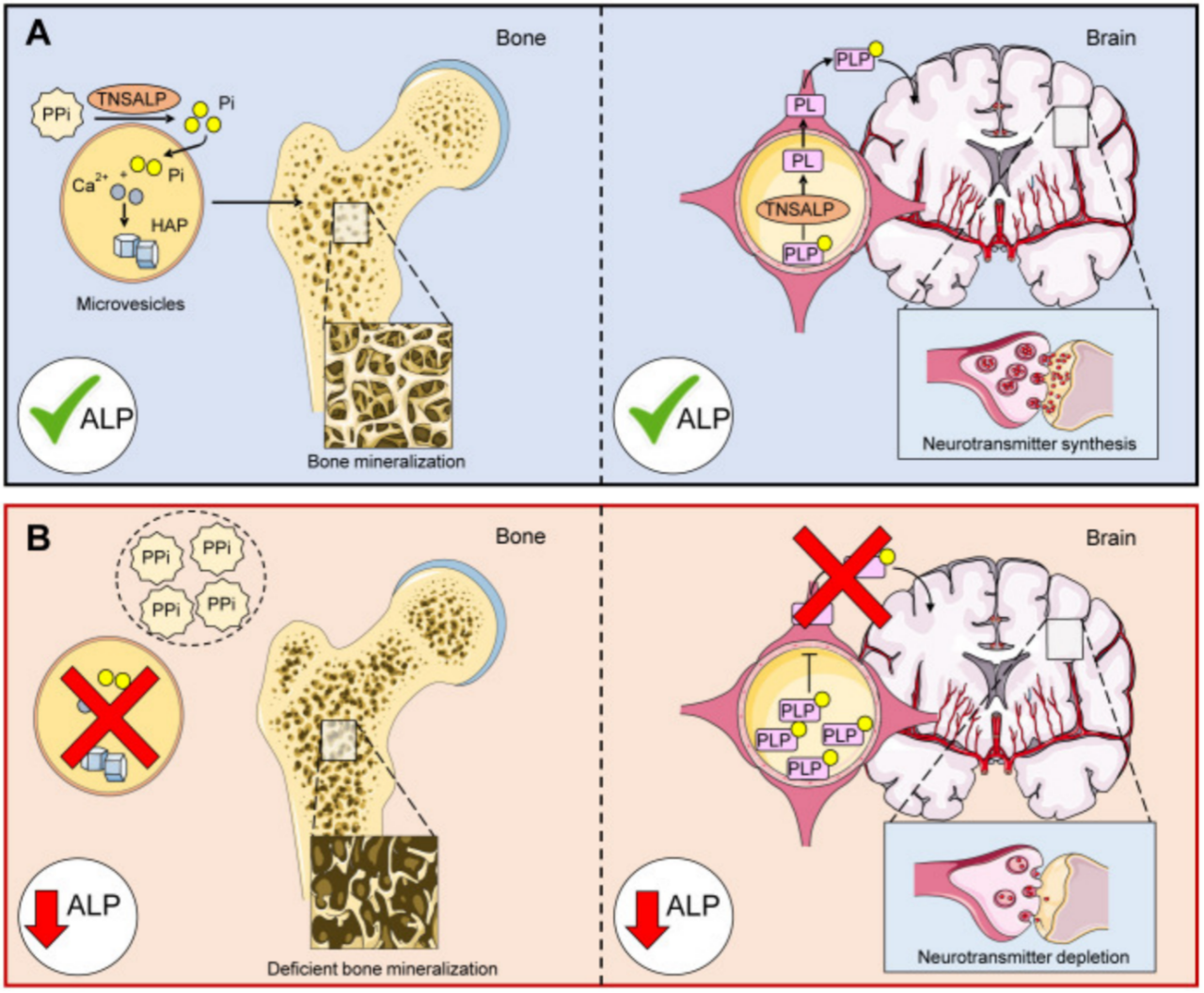
图1. ALP在骨骼矿化及神经递质合成中发挥重要作用(A)ALP正常水平(B)ALP低水平
目前尚无可以完全治愈低碱性磷酸酯酶症的方法,多采用对症治疗。常用的治疗方法为全身治疗、口腔治疗和酶代替治疗方法。其中酶替代治疗方法已经成为低碱性磷酸酯酶症的重要治疗方法。唯一已被FDA批准的酶替代药物Asfotase alfa,治疗费用较高,且需长期用药,对患者造成较大压力。其他新兴疗法如AAV疗法或细胞疗法在研发过程中。而作为罕见病,HPP的发病率仅约为1/100,000,低发病率导致了临床受试者招募困难、药物开发速度受限。
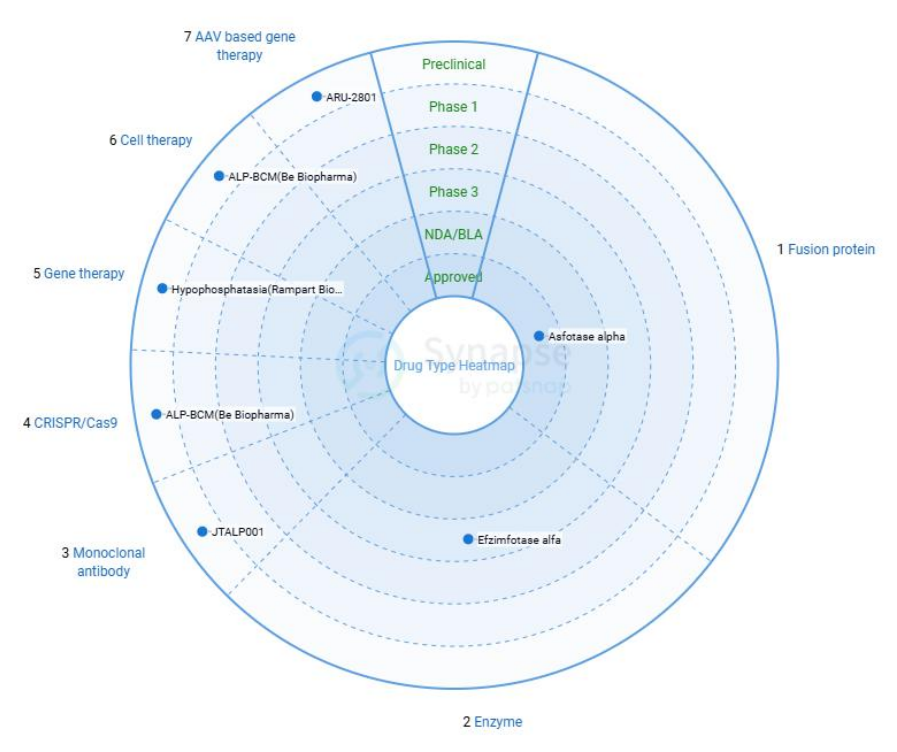
图2. HPP治疗药物开发进展
为助力HPP药物研发,提高药物在临床前早期研究中的评价效率与精准度,集萃药康推出了Alpl-KO小鼠(品系编号:T014130)。该小鼠按照如下图的基因编辑策略,在C57BL/6JGpt小鼠中实现了ALPL基因纯合敲除(KO/KO)。
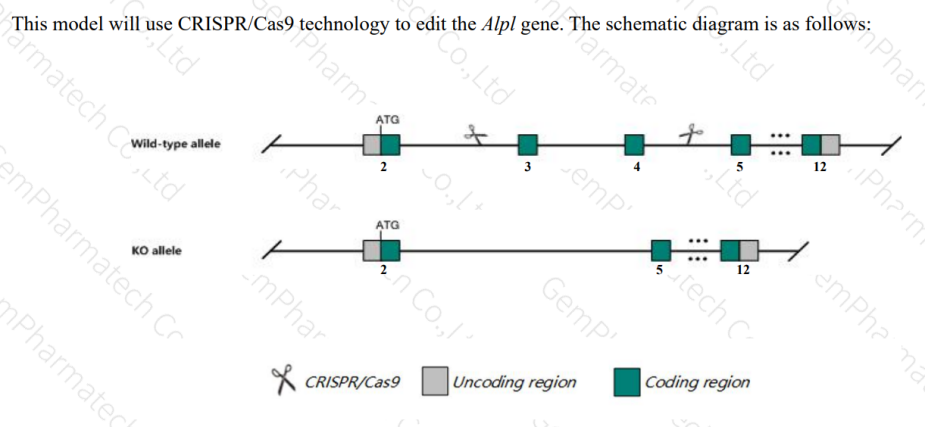
图3. Alpl-KO小鼠基因编辑策略
集萃药康已对该品系小鼠进行了部分关键数据的收集。我们发现,Alpl-KO纯合小鼠如不进行人为干预,在出生后可致死;通过在饮食中补充维生素B6可延长Alpl-KO纯合小鼠的存活期,但仍存在发育迟滞(D21体重显著降低)及断奶前部分致死的表型。
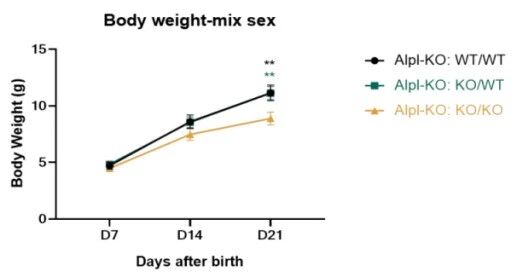
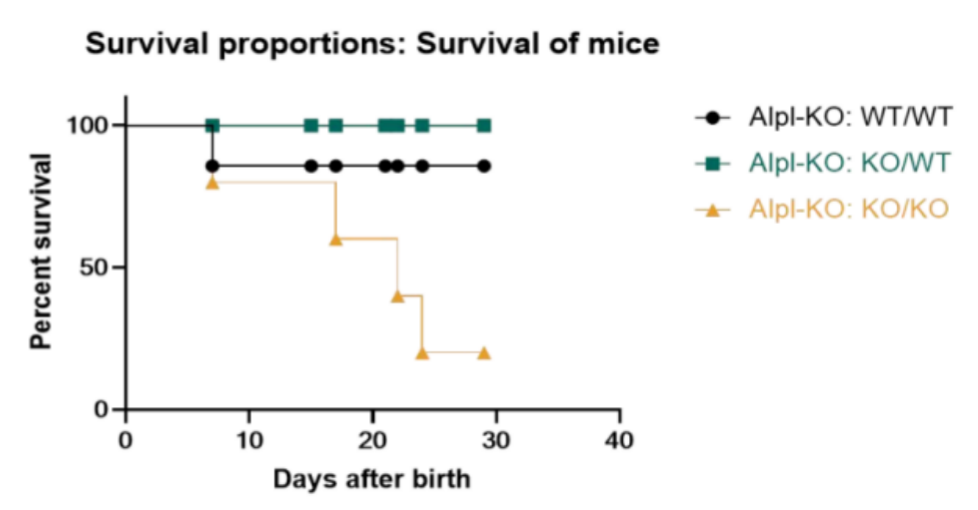
图4. Alpl-KO小鼠体重与生存率情况
左图:体重;右图:生存率;纯合:Alpl-KO:KO/KO,杂合:Alpl-KO:KO/WT,野生:Alpl-KO:WT/WT。样本量:n=15 (Alpl-KO:KO/KO D7)→n=12 (Alpl-KO:KO/KO D14)→n=9 (Alpl-KO:KO/KO D21);n=10 (Alpl-KO:KO/WT);n=10 (Alpl-KO:WT/WT)
对D21时的Alpl-KO纯合小鼠的血清ALP活性与磷酸盐水平检测结果表明,即使经历了维生素B6干预,Alpl-KO纯合小鼠的血清ALP活性与磷酸盐水平在也相较于杂合小鼠或野生小鼠显著降低,如下图所示。
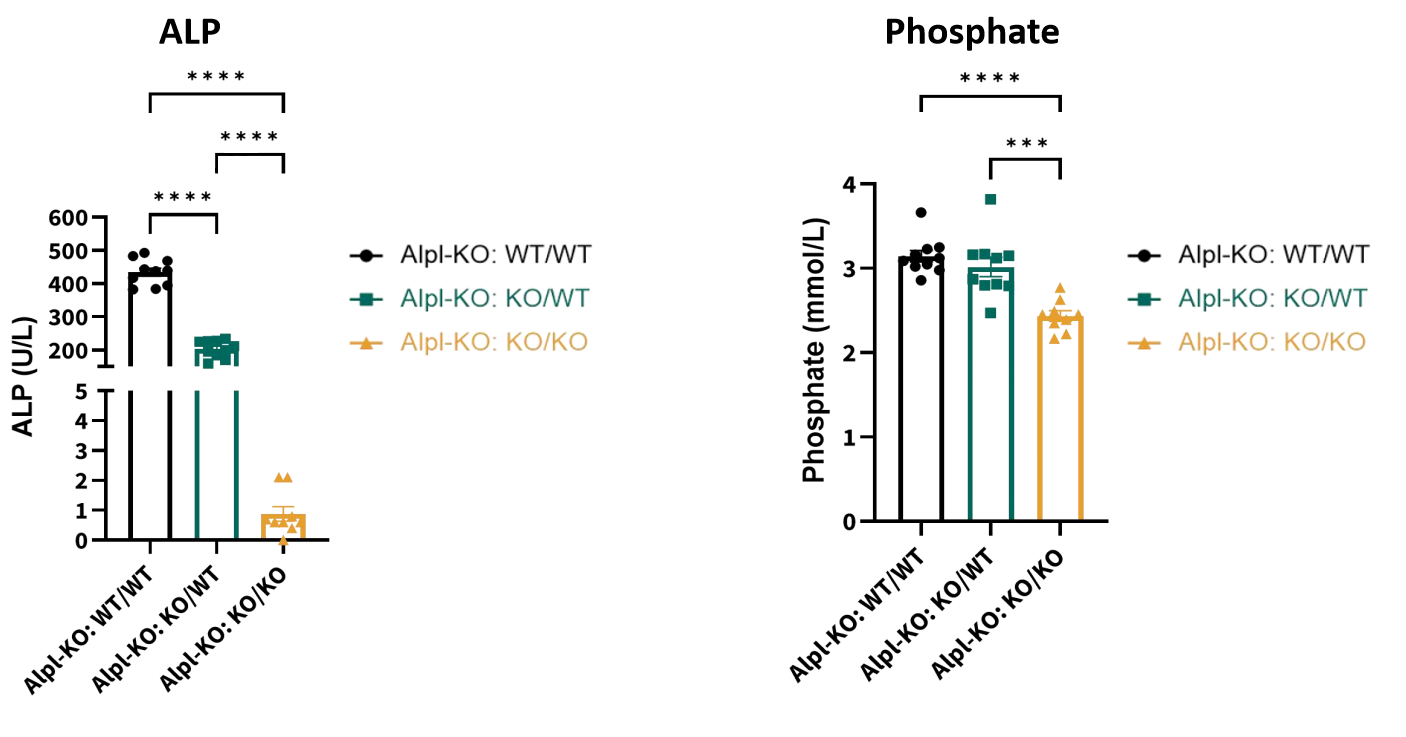
图5. Alpl-KO小鼠血清ALP活性与磷酸盐水平
左图:血清ALP活性;右图:磷酸盐水平。纯合:Alpl-KO:KO/KO,杂合:Alpl-KO:KO/WT,野生:Alpl-KO:WT/WT。样本量:n=9 (Alpl-KO:KO/KO);n=10 (Alpl-KO:KO/WT);n=10 (Alpl-KO:WT/WT)
与HPP患者的临床表现类似,Alpl-KO纯合小鼠的骨骼矿化情况同样存在异常,在D8-D22的各时间DEXA检测(双能X射线吸收测量法)均发现,Alpl-KO纯合小鼠的BMD(骨矿物质密度)、BMC(骨矿物质含量)相较于杂合或野生小鼠有所下降,如下图所示。
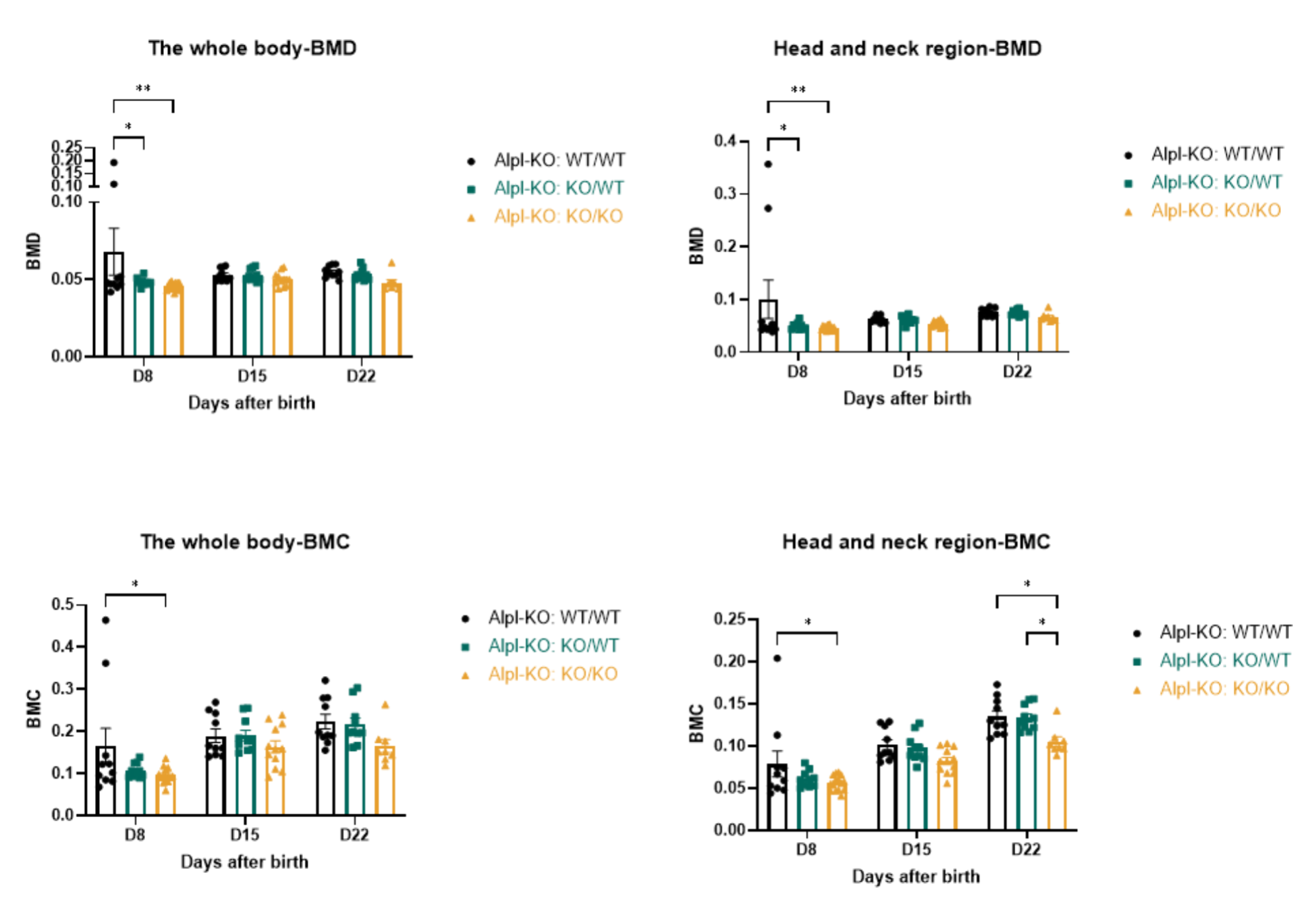
图6. Alpl-KO小鼠BMD及BMC检测结果
纯合:Alpl-KO:KO/KO,杂合:Alpl-KO:KO/WT,野生:Alpl-KO:WT/WT。样本量:n=15 (Alpl-KO:KO/KO D8)→n=12 (Alpl-KO:KO/KO D15)→n=8 (Alpl-KO:KO/KO D22);n=10 (Alpl-KO:KO/WT);n=10 (Alpl-KO:WT/WT)
Alpl-KO小鼠模型通过完全敲除ALPL基因,精准模拟了人类低磷酸酯酶症(HPP)的核心病理机制,能够出现骨骼矿化障碍、新生儿致死的典型表型。Alpl-KO小鼠通过人为补充维生素B6可部分延长生存期,为机制研究及新型靶向药物的疗效评价提供了关键时间窗口,成为了可填补重症HPP临床前研究的工具空白的关键模型。