

疾病罕见爱常存——集萃药康带你了解罕见病之黏多糖贮积症
黏多糖贮积症(mucopolysaccharidosis,MPS)是一组复杂的、进行性多系统受累的溶酶体贮积病,是由于降解糖胺聚糖(亦称酸性黏多糖,glycosaminoglycan,GAGs)的酶缺乏所致。黏多糖是一类复杂的直链多糖,在正常情况下,它们在细胞内被分解和代谢;不能完全降解的黏多糖会在溶酶体中贮积,可造成包括发育迟缓、骨骼畸形、面容异常、神经系统受累、心脏病变、角膜混浊等疾病表型。目前中国大陆缺乏黏多糖贮积症的流行病学资料,但该病已被纳入中国《第 一批罕见病目录》。黏多糖贮积症可根据不同基因编码的不同溶酶体类型将其分为7个大类,11种亚型[1](表1),除MPS Ⅱ型为X连锁遗传外,其余皆属常染色体隐性遗传。
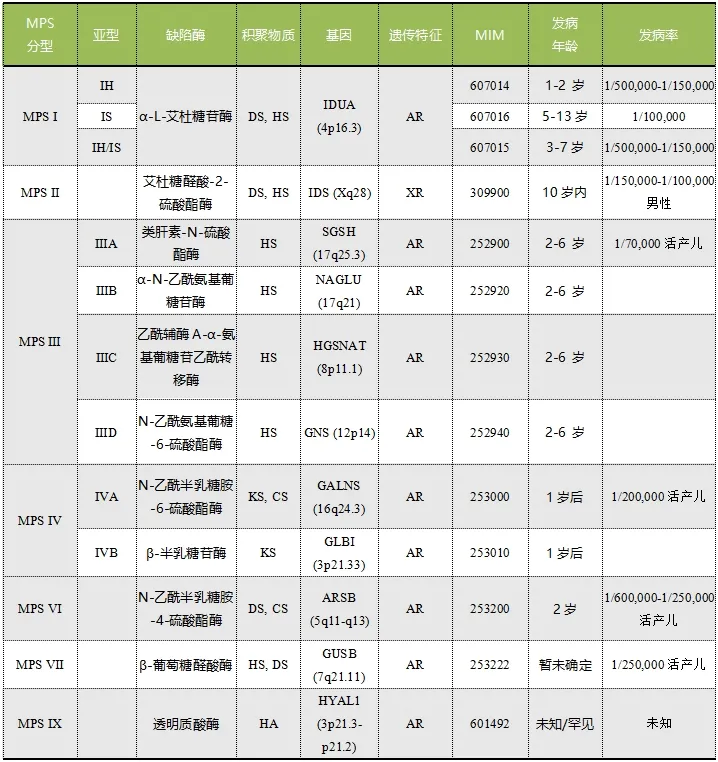
注:HS,硫酸乙酰肝素;DS,硫酸皮肤素;CS,硫酸软骨素;KS,硫酸角质素;HA,透明质酸;AR,常染色体隐性遗传;XR,X染色体隐性遗传。
表1.MPS分型
黏多糖贮积症药物研发现状
当前针对MPS患者采用的治疗方式有酶替代疗法(enzyme replacement therapy,ERT)、造血干细胞移植(hematopoietic stem cell transplantation,HSCT)、底物减少疗法(substrate reduction therapy,SRT)和各种外科手术。国外上市药物主要为酶替代治疗药物,如Aldurazyme(MPS Ⅰ),hunterase和elaprase(MPS Ⅱ), vimizim(MPS ⅣA),naglazyme(MPS Ⅵ)和mepseⅦ(MPS Ⅶ)。但上述疗法旨在减少非神经系统症状和疼痛,无法做到完全治愈MPS患者。近年来基因治疗逐渐火热,通过修复酶缺陷的以期达到一次性永久性治疗的目的。在过去的三十年里,关于MPS的新兴研究方向和治疗方法也主要围绕基因治疗展开,MPS基因治疗的临床前研究数量逐年增加,针对某些类型MPS的基因治疗临床试验正在美国、澳大利亚和一些欧洲国家进行[2-3]。
集萃药康MPS小鼠模型助力临床前研究
目前MPS新药研发仍具有巨大的潜力,在研发过程中离不开合适动物模型的加持。针对MPS不同分型的发病机制及对应的缺陷基因,集萃药康利用基因编辑技术构建了众多MPS小鼠模型。
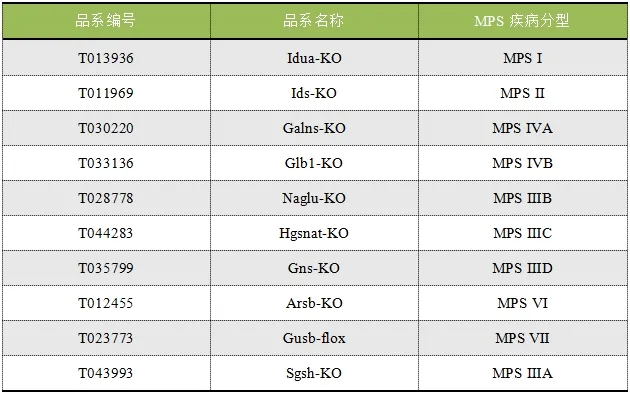
表2.MPS小鼠模型列表
部分小鼠模型数据展示
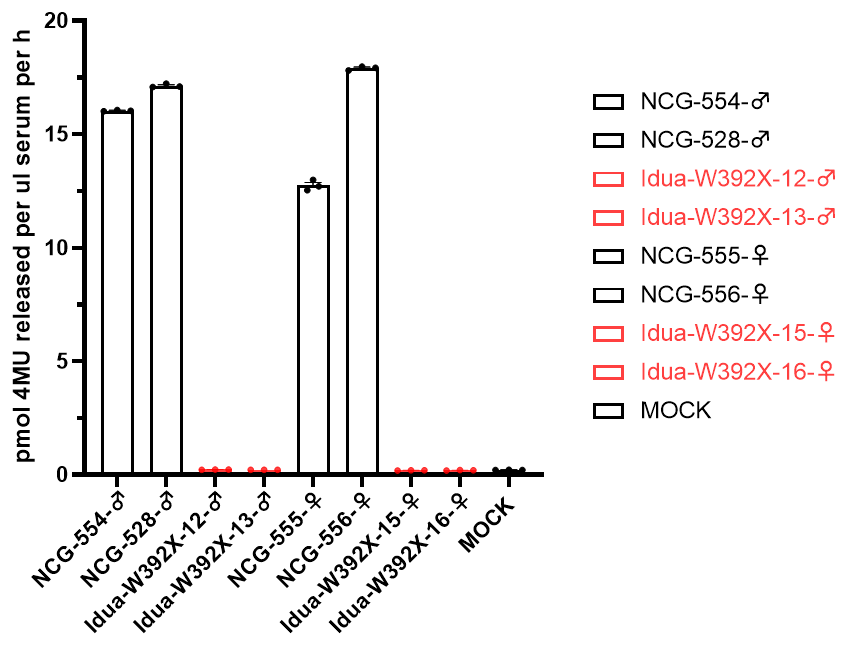
图1.NCG-Idua W392X小鼠血清酶活显著降低
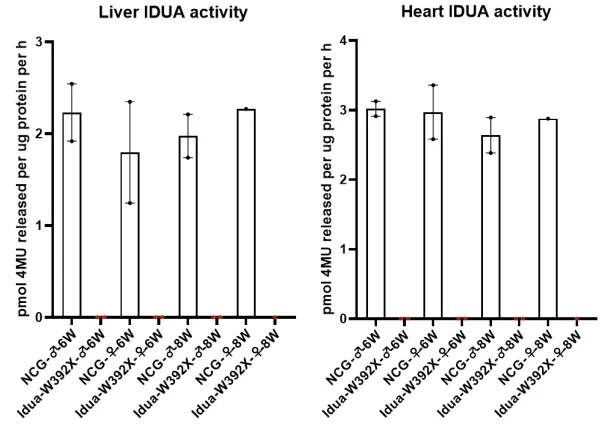
图2.NCG-Idua W392X小鼠肝脏和心脏组织酶活显著降低
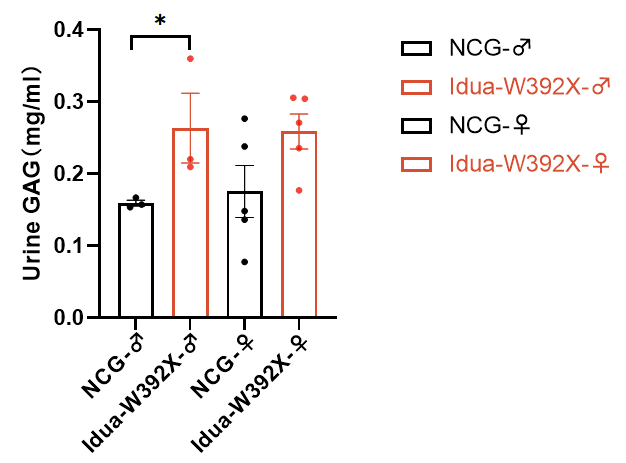
图3.NCG-Idua W392X雄鼠尿液GAG含量显著升高
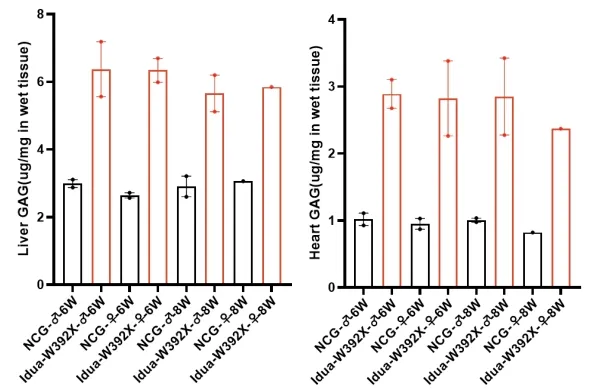
图4.NCG-Idua W392X小鼠肝脏和心脏组织GAG含量升高
参考文献
[1] Bhakthaganesh K, et al. Mucopolysaccharidosis. Taiwan J Ophthalmol. 2023;13(4):443-450.
[2] Ago Y, et al. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int J Mol Sci. 2024;25(2):1113.
[3] Sawamoto K, et al. Gene therapy for Mucopolysaccharidoses. Mol Genet Metab. 2018;123(2):59-68.