

疾病罕见爱常存——集萃药康带你了解罕见病之肝豆状核变性
肝豆状核变性简介
肝豆状核变性(hepatolenticular degeneration,HLD),又名Wilson病(Wilson disease,WD),是一种由于铜转运ATP酶β(ATPase copper transporting beta,ATP7B)基因突变而导致的遗传性铜代谢障碍疾病。Kinnier Wilson在1912年首次描述此病后,人们发现肝脏是全身铜代谢的活跃器官,膳食铜通过门静脉转运并贮存在肝脏中。ATP7B编码跨膜铜转运ATP酶,其功能障碍导致铜胆汁排泄途径受损,铜在肝脏中逐渐积累。当肝脏储存铜的能力耗竭时,过量的非铜蓝蛋白结合的铜进入体循环并沉积在各个器官中,产生肝外铜毒性,特别是在大脑中沉积,可导致神经症状和精神障碍。WD可在任何年龄发病,主要以儿童、青少年多见[1],我国尚缺乏全国性流行病学调查资料,推测患病率为0.587/10000[2]。
肝豆状核变性药物研发现状
WD是目前可用药物治疗的遗传代谢性疾病之一,治疗越早,预后越好。WD的治疗药物可以分为两类,一类为铜螯合剂,增加尿铜排泄,如D-青霉胺等;另一类则为以锌剂为代表的减少铜吸收的药物。此外还有肝移植及低铜饮食等方式可以控制疾病的发生发展[3]。但单纯的抗铜治疗无法修正患者的基因缺陷,需要终身进行维持治疗,因此基因治疗是实现根治肝豆状核变性的最 佳方法。Vivet Therapeutics SAS公司生产的VTX-801是一种新型重组腺相关病毒基因治疗载体,提供一种微型化ATP7B转基因编码功能蛋白,恢复铜稳态,目前正处于临床1/2期试验[4]。
集萃药康Atp7b-KO小鼠模型助力临床前研究
目前关于肝豆状核变性发病的机制机理仍不明确,新药研发也具有巨大的潜力。小鼠模型由于和人类在生理、病理和遗传学方面有很多相似之处,为病理机制的研究、药物的筛选、优化和安全性评估提供了重要的手段和依据。集萃药康采用基因编辑技术,对C57BL/6JGpt小鼠的Atp7b基因进行编辑,得到Atp7b基因敲除小鼠模型,纯合子小鼠出现铜代谢异常表型。
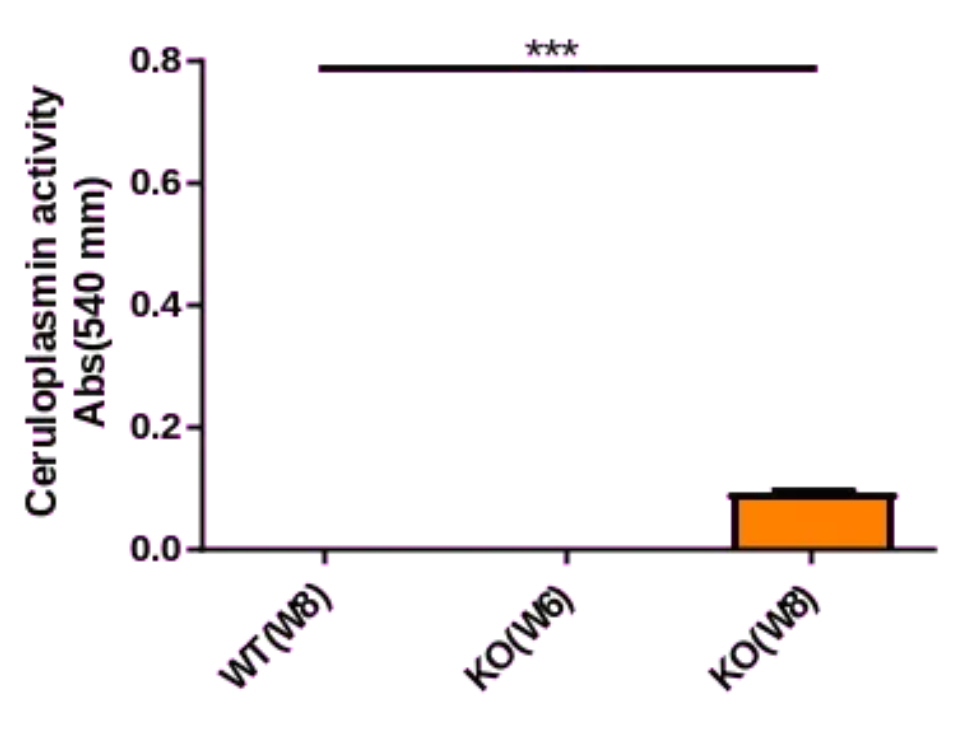
图1. Atp7b-KO小鼠血浆铜蓝蛋白活性检测
通过酶活性法检测血浆铜蓝蛋白活性,结果显示KO小鼠中的铜蓝蛋白活性显著降低。
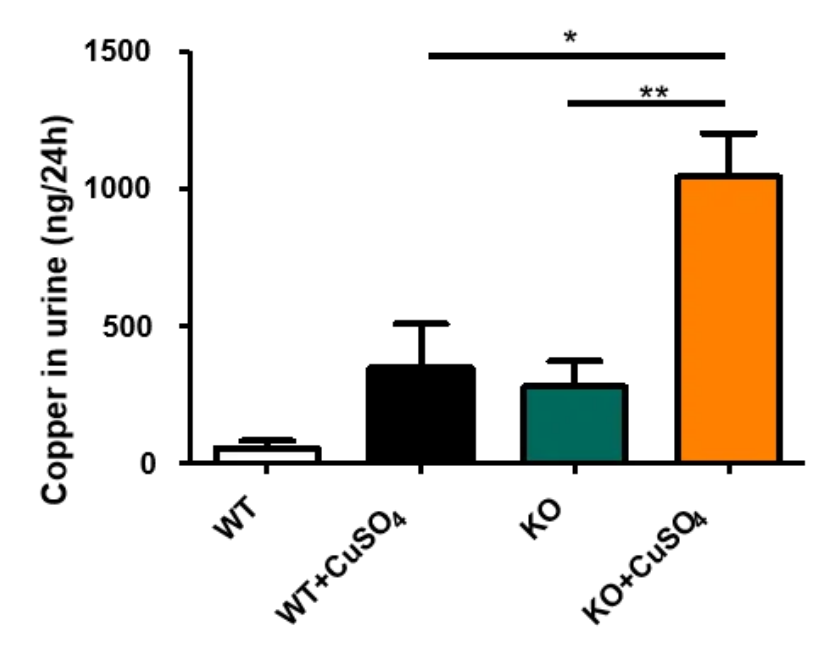
图2. Atp7b-KO小鼠尿液铜含量检测
小鼠尿液铜含量检测结果显示,本底水平,ATP7b-KO小鼠尿铜含量略高于野生型小鼠,腹腔注射CuSO4后,ATP7b-KO小鼠尿液铜含量显著高于野生型小鼠。
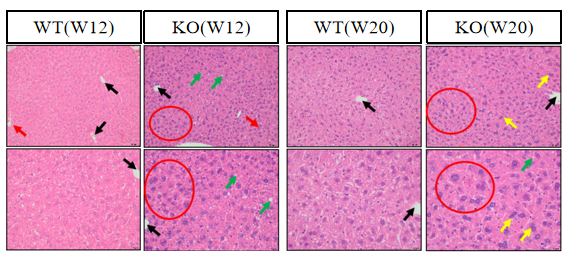
图3. Atp7b-KO小鼠肝脏病理形态
HE染色检测肝脏组织形态和病变情况。结果显示,部分Atp7b-KO小鼠肝脏产生病变,出现肝脏细胞脂肪变性、细胞肥大等异常表型。(中央静脉(黑色箭头);小胆管(红色箭头);脂肪变性(绿色箭头);肝细胞肥大(红色圆圈);双核/多核肝细胞(黄色箭头)。)
参考文献
[1] Nagral A, Sarma MS, Matthai J, et al. Wilson's disease: clinical practice guidelines of the indian national association for study of the liver, the Indian Society of Pediatric Gastroenterology,
Hepatology and Nutrition, and the Movement Disorders Society of India[J]. J Clin Exp Hepatol, 2019, 9(1): 74-98. DOI: 10.1016/ j.jceh.2018.08.009.
[2] Xie JJ, Wu ZY. Wilson's disease in China[J]. Neurosci Bull, 2017, 33(3): 323-330. DOI: 10.1007/s12264-017-0107-4.
[3]肝豆状核变性诊疗指南(2022年版).中华肝脏病杂志, 2022, 30(1) : 9-20.
[4]https://www.vivet-therapeutics.com/vtx%e2%80%90801-receives-u-s-fda-fast-track-designation-for-the-treatment-of-wilson-disease/